
Isomers hosting three epoxides prefer axial orientation by a huge margin (>40kcal/mol) despite having severe steric problems. Among the two axial isomers, the ‘all-cis’ structure 1B is less stable than the cis-trans hy-brid 1A though the C-C bond alternation is moderate in 1B compared to 1A. This reduced bond alternation in 1B along with the fact that it also has a planar cyclohexane ring unlike 1A suggests the possibility of cyclic conjuga-tion à la aromatic delocalization in 1B; however, it is not dominating the energetics compared to the repulsive O…..O interactions. The splaying of cis oxygen atoms that adversely affect C-C bonding is less in 1B (0.600Å) than 1A (0.624Å), facilitating cyclic delocalization in ‘all-cis’ form.

Overlap Control of Conformations
Bonding in epoxides is usually explained by two classical models, though both have their own drawbacks. The popular bent ‘banana bond’ model of Coulson is borrowed from cyclopropane that uses isovalent hybridization. Coulson attributes shortening of exo-epoxide bonds to the increased s character but the absence of expected elongation of epoxide C-C bond forced him to assert that these bonds are bent outwards.

The alternate Walsh MO
model assumes sp2 hybridization for the carbon41 due to its propensity for addition like olefins, where the
shortening of exo-epoxy bonds is attributed to conjugation. In all the epoxide dimers, the central C-C
bond is ~0.05Å shorter than standard C-C single bond, except for the transition
state 2B which still experiences
steric repulsion from oxygen lone-pairs.
This shortening is found even in 2F
despite the fact that their epoxides are oriented orthogonally indicating that
conjugation advocated by the Walsh model may not be the reason. Similarly, the destabilization of 2E that has trans orientation of epoxides is hard to account for with the Walsh
model of isovalent hybridization. It is clear that these simple models by
Coulson and Walsh are not adequate to explain the energetic and C-C bond length
variations of different conformations of epoxide dimers.

Analysis
of the interaction of frontier MOS of epoxide fragments to form 1,3 trans inter and intra epoxide dimers show
negligible conjugative stabilization due to the large HOMO-LUMO gap of epoxides
as expected. Though the diagrasm also
shows inter to be more stable than intra as observed in nanosheets, its MO
origins are difficult to trace from their fragment MOs. This is mostly due to the low symmetry of the
fragments forced by ipso hydrogen atoms of the dimer that allows the seamless
mixing of many frontier MOs.

To avoid this complication, we use the
frontier MOs of the epoxide fragment when two geminal hydrogens are removed
from the carbon atom to construct the fragment MOs. This gives
two MOs dominated by the divalent carbon in the frontier; one is an sp hybrid pointing
outwards and another p-type orbital perpendicular to the epoxide ring. The orientation of these two types of FMOs (sp
and p) forming the exo-C-C bond in different environments of the dimer is given
in Figure 3.
In the inter case, the dominant interaction
is between the sp hybrid of one ipso carbon with the p orbital of the
other. Among these, trans has symmetrical distribution around the exo-epoxy bonds, while
cis has electron density bent away
from the epoxide bridges. In the intra
case, p-p and sp-sp overlap are dominant. Among these two, the overlap between
sp hybrids in trans is poor as they
are pointing away, while in cis they
have the best overlap. The variation in
the bond-lengths of the C2O nanosheets, particularly the computed
energetic proximity of 3A and 3B nanosheets, supports the above
reasoning. Despite splaying, 3B shows the shortest C-O distances
indicating that splaying is not detrimental to the stability of the carbon
framework as it helps improve sp-p overlap in the exo C-C region. This reasoning implies the bending of both
epoxy and exo-epoxy bonds and can potentially be used to predict the mode and
ease of nucleophilic attack.

These facts naturally lead to a new model of epoxide bonding based on sp hybridization, in which s orbital and the radial p orbital of the epoxide ring is mixed to yield one sp hybrid pointing inwards and another pointing outward. The inward-pointing sp hybrid, along with the p orbital tangential to the epoxide ring, is responsible for the bonding within the epoxide ring while the outward-pointing sp hybrid with the p orbital perpendicular to the epoxide ring is presumed to be involved in the exo-epoxy bonding. This new hybridization model based on FMO theory is similar to sp hybrid model typically used in polyhedral boranes except that only one of the unhybridized p orbital (tangential to the epoxide ring) is involved in the bonding within the epoxide ring while the unhybridized p orbital perpendicular to the ring is engaged in exo-epoxy bonding.
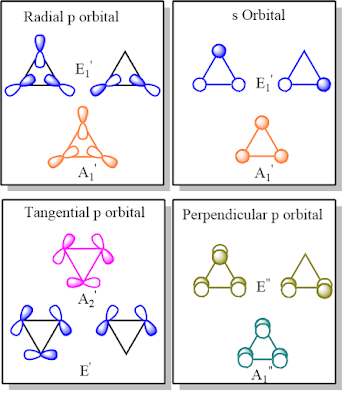
This model successfully addresses
the shortcomings of the Walsh model, doing away with the conjugative
stabilization as the reason for bond shortening. It rather attributes to the overlap of the
frontier orbitals away from the internuclear line making the exo-epoxy bonds
also to have substantial electron density away from the inter-nuclear region,
effectively extending the idea of bending to cover exo-epoxy bonds. Upon extension,
the sp hybridization model can also explain the perceived instability of four-membered
rings over three-membered rings as arising from repulsive 1,3 interactions
between inward-pointing sp hybrids and will be described elsewhere. With the observed trends in energetics and
geometry successfully explained, our results provide the framework for understanding
the origins of many of the mysterious observations surrounding the graphene
oxide structure.
https://link.growkudos.com/1qinwskowsg
Comments
Post a Comment